


As mitocôndrias são conhecidas como potências das células, mas evidências crescentes sugerem que elas também desempenham um papel na inflamação. Cientistas do Salk Institute e da UC San Diego publicaram novas descobertas na Immunity em 26 de julho de 2022, onde examinaram células sanguíneas humanas e descobriram uma ligação surpreendente entre mitocôndrias, inflamação e DNMT3A e TET2 – dois genes que normalmente ajudam a regular o crescimento das células sanguíneas, mas , quando mutados, estão associados a um risco aumentado de aterosclerose.
“Descobrimos que os genes DNMT3A e TET2, além de seu trabalho normal de alterar marcadores químicos para regular o DNA, ativam diretamente a expressão de um gene envolvido nas vias inflamatórias mitocondriais, o que sugere um novo alvo molecular para a terapêutica da aterosclerose”, diz Gerald Shadel, co-autor sênior, professor Salk e diretor do San Diego Nathan Shock Center of Excellence in the Basic Biology of Aging.
O estudo começou quando pesquisadores da UC San Diego notaram uma resposta inflamatória específica ao investigar os papéis das mutações DNMT3A e TET2 na hematopoiese clonal – quando células sanguíneas imaturas mutadas dão origem a uma população de células sanguíneas maduras com mutações idênticas. Eles relataram que a sinalização inflamatória anormal também estava relacionada à deficiência de DNMT3A e TET2 nas células sanguíneas que desempenham um papel importante na resposta inflamatória que promove a progressão da aterosclerose.
Mas como os genes DNMT3A e TET2 estavam envolvidos na inflamação e, possivelmente, na aterosclerose, era desconhecido.
“O problema é que não conseguimos descobrir como o DNMT3A e o TET2 estavam envolvidos porque as proteínas que eles codificam fazem coisas aparentemente opostas em relação à regulação do DNA”, diz Christopher Glass, co-autor sênior e professor da UC San Diego School of Medicine. “Sua atividade antagônica nos levou a acreditar que pode haver outros mecanismos em jogo. Isso nos levou a adotar uma abordagem diferente e entrar em contato com Shadel, que havia descoberto a mesma via inflamatória anos antes enquanto examinava as respostas ao estresse do DNA mitocondrial”.
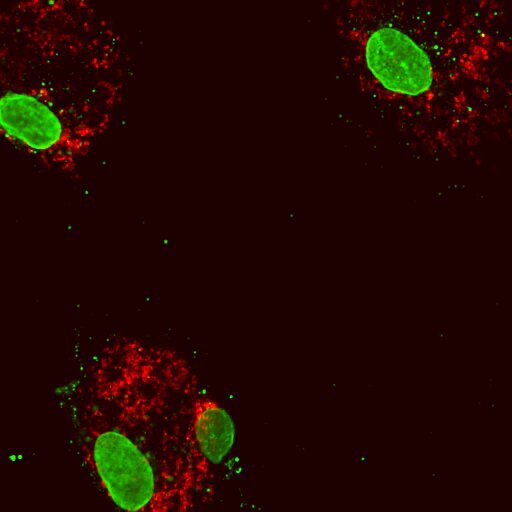
Dentro das mitocôndrias reside um subconjunto único do DNA da célula que deve ser organizado e condensado corretamente para sustentar a função normal. A equipe de Shadel investigou anteriormente os efeitos do estresse do DNA mitocondrial removendo o TFAM, um gene que ajuda a garantir que o DNA mitocondrial seja empacotado corretamente. Eles descobriram que quando os níveis de TFAM são reduzidos, o DNA mitocondrial é expelido das mitocôndrias para o interior da célula. Isso dispara o mesmo alarme molecular que informa à célula que há um invasor bacteriano ou viral e desencadeia uma via molecular defensiva que promove a inflamação.
Cientistas dos laboratórios Glass e Shadel trabalharam juntos para entender melhor por que as mutações DNMT3A e TET2 levaram a respostas inflamatórias semelhantes às observadas durante o estresse do DNA mitocondrial. As equipes aplicaram ferramentas de engenharia genética e imagens de células para examinar células de pessoas com células normais, aquelas com mutações de perda de função na expressão de DNMT3A ou TET2 e aquelas com aterosclerose.
Eles descobriram que reduzir experimentalmente a expressão de DNMT3A ou TET2 nas células sanguíneas normais teve resultados semelhantes às células sanguíneas que tinham mutações de perda de função e células sanguíneas de pacientes com aterosclerose – um aumento da resposta inflamatória. Notavelmente, baixos níveis de expressão de DNMT3A e TET2 nas células sanguíneas levam à redução da expressão de TFAM, que por sua vez leva ao empacotamento anormal do DNA mitocondrial, instigando a inflamação devido ao DNA mitocondrial liberado.
“Descobrimos que as mutações DNMT3A e TET2 impedem sua capacidade de ligar e ativar o gene TFAM”, diz o primeiro autor Isidoro Cobo, pesquisador de pós-doutorado no laboratório Glass na UC San Diego. “Perder ou reduzir essa atividade de ligação leva à liberação de DNA mitocondrial e a uma resposta hiperativa de inflamação mitocondrial, e acreditamos que isso pode exacerbar o acúmulo de placas na aterosclerose”.
“É muito emocionante ver que nossa descoberta sobre a depleção de TFAM, causando estresse e inflamação no DNA mitocondrial, agora tem relevância direta para uma doença como a aterosclerose”, diz Shadel, que detém a cadeira Audrey Geisel em Ciências Biomédicas. “Desde que revelamos essa via, houve uma explosão de interesse nas mitocôndrias envolvidas na inflamação e muitos relatos ligando a liberação de DNA mitocondrial a outros contextos clínicos”.
As terapêuticas que visam as vias de sinalização da inflamação já existem para muitas outras doenças. Glass e Shadel acreditam que vias de bloqueio que exacerbam a aterosclerose em pacientes com mutações TET2A e DNMT3A podem formar a base para novos tratamentos. Em seguida, os cientistas continuarão investigando esse caminho e investigarão como o DNA mitocondrial está envolvido em outras doenças humanas e no envelhecimento.
Publicado em 23/08/2022 11h36
Artigo original:
Estudo original: