


Os pesquisadores adicionaram 200 milhões de pares de bases de DNA e 115 genes codificadores de proteínas – mas eles ainda precisam sequenciar inteiramente o cromossomo Y.
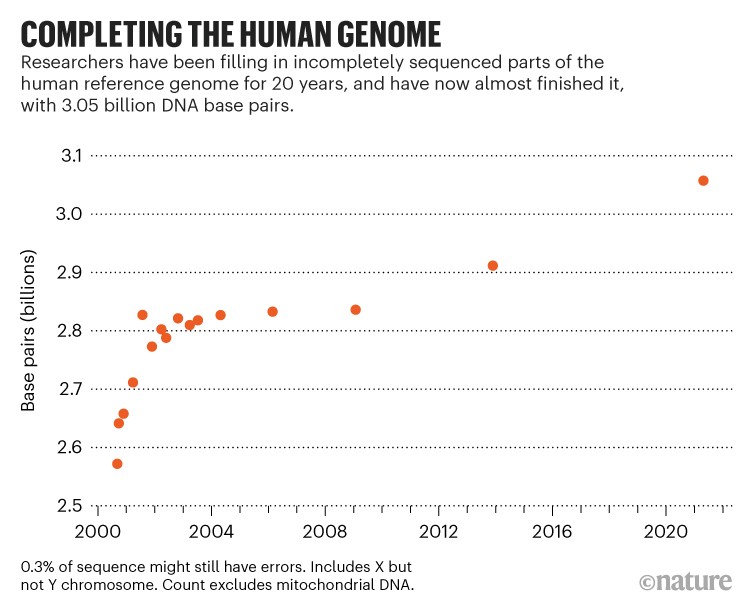
Quando o sequenciamento do genoma humano foi anunciado há duas décadas pelo Projeto Genoma Humano e pela empresa de biotecnologia Celera Genomics, a sequência não estava realmente completa. Faltavam cerca de 15%: as limitações tecnológicas deixaram os pesquisadores incapazes de descobrir como certos trechos de DNA se encaixavam, especialmente aqueles em que havia muitas letras repetidas (ou pares de bases). Os cientistas resolveram parte do quebra-cabeça ao longo do tempo, mas o genoma humano mais recente, que os geneticistas usam como referência desde 2013, ainda carece de 8% da sequência completa.
Agora, os pesquisadores do Consórcio Telomere-to-Telomere (T2T), uma colaboração internacional que compreende cerca de 30 instituições, preencheram essas lacunas. Em um preprint1 de 27 de maio intitulado ‘A sequência completa de um genoma humano’, a pesquisadora de genômica Karen Miga da Universidade da Califórnia, Santa Cruz, e seus colegas relataram que sequenciaram o restante, no processo de descoberta de cerca de 115 novos genes que código para proteínas, para um total de 19.969.
“É emocionante ter alguma solução para as áreas problemáticas”, diz Kim Pruitt, bioinformática do Centro Nacional de Informações sobre Biotecnologia dos EUA em Bethesda, Maryland, que considera o resultado um “marco significativo”.
Nova tecnologia de sequenciamento
O genoma recém-sequenciado – denominado T2T-CHM13 – adiciona quase 200 milhões de pares de bases à versão de 2013 da sequência do genoma humano.
Desta vez, em vez de pegar DNA de uma pessoa viva, os pesquisadores usaram uma linha celular derivada do que é conhecido como uma mola hidatiforme completa, um tipo de tecido que se forma em humanos quando um espermatozoide insemina um óvulo sem núcleo. A célula resultante contém cromossomos apenas do pai, então os pesquisadores não precisam distinguir entre dois conjuntos de cromossomos de pessoas diferentes.
Miga diz que a façanha provavelmente não teria sido possível sem a nova tecnologia de sequenciamento da Pacific Biosciences em Menlo Park, Califórnia, que usa lasers para escanear longos trechos de DNA isolado de células – até 20.000 pares de bases por vez. Os métodos convencionais de sequenciamento leem o DNA em pedaços de apenas algumas centenas de pares de bases por vez, e os pesquisadores remontam esses trechos como peças de um quebra-cabeça. As peças maiores são muito mais fáceis de juntar, porque é mais provável que contenham sequências que se sobrepõem.
T2T-CHM13 não é a última palavra no genoma humano, no entanto. A equipe T2T teve problemas para resolver algumas regiões nos cromossomos e estima que cerca de 0,3% do genoma pode conter erros. Não há lacunas, mas Miga diz que as verificações de controle de qualidade têm se mostrado difíceis nessas áreas. E a célula espermática que formou a mola hidatiforme carregava um cromossomo X, então os pesquisadores ainda não sequenciaram um cromossomo Y, que normalmente desencadeia o desenvolvimento biológico masculino.
Centenas de genomas a seguir
T2T-CHM13 representa o genoma de apenas uma pessoa. Mas o T2T Consortium se associou a um grupo chamado Human Pangenome Reference Consortium, que pretende nos próximos 3 anos sequenciar mais de 300 genomas de pessoas em todo o mundo. Miga afirma que as equipes poderão usar o T2T-CHM13 como referência para entender quais partes do genoma tendem a diferir entre os indivíduos. Eles também planejam sequenciar um genoma inteiro que contém cromossomos de ambos os pais, e o grupo de Miga tem trabalhado no sequenciamento do cromossomo Y, usando os mesmos novos métodos para ajudar a preencher as lacunas.
Miga espera que os pesquisadores de genética descubram rapidamente se alguma das áreas recentemente sequenciadas e possíveis genes estão associados a doenças humanas. “Quando o genoma humano foi lançado, não tínhamos as ferramentas prontas para funcionar”, diz ela, mas as informações sobre a função dos genes recém-sequenciados devem vir muito mais rápido agora, porque “construímos uma tonelada de recursos”.
Ela espera que as futuras sequências do genoma humano abranjam tudo, incluindo as seções recém-sequenciadas – não apenas as partes que são fáceis de ler. Isso deve ser mais fácil agora que o genoma de referência foi concluído e alguns dos empecilhos técnicos foram resolvidos. “Precisamos alcançar um novo padrão em genômica onde isso não seja especial, mas rotineiro”, diz ela.
Publicado em 05/06/2021 18h02
Artigo original:
Estudo original: