


Uma forma previamente desconhecida de uma doença neurodegenerativa grave e progressiva que geralmente afeta adultos mais velhos foi identificada em crianças a partir dos três anos de idade.
A esclerose lateral amiotrófica (ELA) é um distúrbio neurológico raro em que a degeneração do neurônio motor leva a sérios prejuízos no movimento muscular voluntário.
A condição, que causa fraqueza cada vez maior nos músculos de todo o corpo, torna difícil caminhar, falar e, eventualmente, até respirar, levando à morte na maioria dos pacientes alguns anos após a manifestação dos sintomas.
A maioria dos casos de ELA surge em pessoas com idade entre 55 e 75 anos, e a maioria dos casos é considerada esporádica, com a causa permanecendo desconhecida.
Para algumas pessoas, no entanto, a doença se apresenta de maneira muito diferente. Em uma minoria de casos, a genética parece desempenhar um papel causal – e às vezes a doença aparece em pessoas muito mais jovens.
Em um novo estudo, os cientistas descobriram que ambas as manifestações mais raras da doença coincidiam, descobrindo um conjunto incomum de mutações ligadas a uma forma distinta de ELA genética em crianças, cujas misteriosas doenças causadoras de perda de massa haviam intrigado seus médicos por anos.
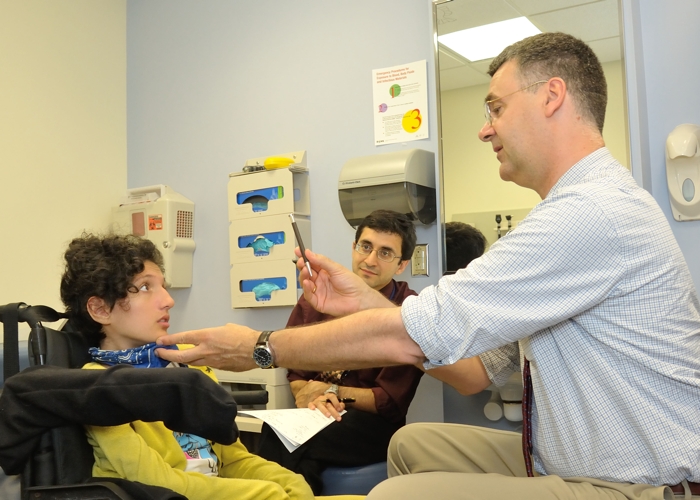
No mais divulgado desses casos dolorosos, uma adolescente italiana chamada Claudia Digregorio acabou se encontrando com o Papa depois que sua doença degenerativa não identificada chamou a atenção no YouTube.
Ao mesmo tempo, cientistas do Instituto Nacional de Doenças Neurológicas e Derrame (NINDS) em Bethesda, Maryland, começaram a investigar a doença de Claudia, na esperança de encontrar respostas para a causa subjacente de sua condição.
Agora, em uma investigação internacional de 10 desses pacientes jovens, muitos dos quais começaram a apresentar sintomas semelhantes aos da ELA na primeira infância, os pesquisadores identificaram uma base genética para este subtipo raro de uma doença já rara.
“Esses pacientes jovens tinham muitos dos problemas do neurônio motor superior e inferior que são indicativos de ELA”, disse o neurologista Payam Mohassel do National Institutes of Health.
“O que tornou esses casos únicos foi a idade precoce de início e a progressão mais lenta dos sintomas. Isso nos fez questionar o que estava por trás dessa forma distinta de ELA”.
A resposta parece estar em um conjunto de mutações em um gene chamado SPTLC1, que codifica uma proteína que atua como um catalisador na produção de moléculas gordurosas chamadas esfingolipídios.
O DNA dos 10 pacientes jovens no estudo revelou mutações no gene SPTLC1, com quatro dos pacientes (todos de uma família) herdando suas variações de um de seus pais, enquanto os outros seis, pacientes não aparentados, pareceram mostrar de novo aleatório mutações (presentes pela primeira vez em um membro da família) no gene.
Em ambos os casos, as mutações são problemáticas, levando à produção excessiva de esfingolipídios, ligados a níveis anormalmente elevados de uma enzima que ajuda a formar os lipídios, chamada serina palmitoiltransferase (SPT).
“Nossos resultados sugerem que esses pacientes com ELA estão essencialmente vivendo sem um freio na atividade do SPT”, disse a bioquímica Teresa M. Dunn da Uniformed Services University.
“O SPT é controlado por um ciclo de feedback … As mutações que esses pacientes carregam basicamente causam um curto-circuito nesse ciclo de feedback.”
Anormalidades na atividade do SPT devido a mutações no gene SPTLC1 foram previamente associadas à neurodegeneração em outra doença, chamada Neuropatia Hereditária Sensorial e Autonômica (HSAN1), embora o mecanismo patológico pareça ser diferente.
Em HSAN1, as mutações SPTLC1 produzem esfingolipídios prejudiciais; no presente estudo, as mutações SPTLC1 encontradas produziram níveis anormais de esfingolipídeos não prejudiciais, ao inibir uma proteína chamada ORMDL de regular a atividade do SPT.
Por sua vez, o excesso de oferta de esfingolipídeos se acumula nos neurônios motores humanos, levando ao início precoce da doença do neurônio motor, que a equipe caracteriza como ‘ELA de início na infância’.
Felizmente, pode haver uma maneira de impedir que isso aconteça, com os pesquisadores descobrindo uma maneira de desligar o gene SPTLC1 mutante usando pequenos RNA interferentes (siRNA): moléculas de RNA que visam especificamente o alelo mutante e inibem a produção de SPT hiperativa .
Embora o experimento só tenha sido testado nas células dos pacientes por enquanto – ainda não nos próprios pacientes – o avanço aponta para um futuro onde os tratamentos podem ser possíveis, quando as crianças podem, esperançosamente, ser poupadas do pior desta doença debilitante e desesperadora.
“Esses resultados preliminares sugerem que podemos ser capazes de usar uma estratégia de silenciamento de gene de precisão para tratar pacientes com este tipo de ELA”, disse o pesquisador sênior do NINDS, Carsten Bönnemann.
“Nosso objetivo final é traduzir essas idéias em tratamentos eficazes para nossos pacientes que atualmente não têm opções terapêuticas.”
Publicado em 01/06/2021 13h36
Artigo original:
Estudo original: