



O caminho para as terapias genéticas para doenças genéticas tem sido longo – e caro – mas o campo poderá receber boas notícias em breve. Em 22 de junho, a Food and Drug Administration dos EUA decidirá se concederá uma aprovação rápida à primeira terapia genética para a distrofia muscular de Duchenne (DMD), uma doença genética que afeta cerca de 1 em 3.500 meninos. Crianças com DMD não conseguem produzir uma proteína chamada distrofina, resultando em degeneração muscular progressiva e morte aos 20 anos devido a insuficiência cardíaca ou respiratória.
A terapia, conhecida como SRP-9001, é feita pela Sarepta Therapeutics, com sede em Cambridge, Massachusetts. Seria a 13ª terapia genética que o FDA aprovou desde 2017 e a primeira a atingir uma doença genética prevalente em crianças. A aprovação acelerada permitiria que a droga chegasse ao mercado antes que grandes ensaios clínicos fossem concluídos, com base em evidências de que a terapia permite que os meninos produzam uma forma modificada de distrofina.
A data da decisão foi adiada no final de maio, depois que funcionários e consultores da FDA levantaram preocupações sobre a força dos dados de Sarepta até agora; O SRP-9001 parece ter apenas um efeito modesto na função muscular e apenas em algumas pessoas. Um comunicado de imprensa da empresa diz que a agência provavelmente o aprovará apenas para meninos de quatro e cinco anos, mas pode expandir esse intervalo se um ensaio clínico em andamento mostrar que a eficácia do medicamento vale os riscos associados a ele. Os analistas preveem que o tratamento único custará US$ 2 milhões. Um porta-voz da Sarepta se recusou a revelar o preço até que o medicamento seja aprovado, mas disse que terá um preço “abaixo do valor que trará aos pacientes”.
Ainda assim, alguns cientistas esperam que a aprovação abra caminho para mais terapias genéticas. “Ter um grande sucesso como este vai validar bastante a tecnologia”, diz Jeffrey Chamberlain, neurologista da Universidade de Washington em Seattle, que desenvolveu algumas das tecnologias usadas pelo Sarepta. “É uma maneira de colocar o pé na porta agora e ajudar as crianças.”
Por que a DMD tem sido tão difícil de tratar?
DMD parece ser um alvo direto para a terapia genética. A doença atinge quase exclusivamente meninos, pois eles possuem apenas uma cópia do cromossomo X, onde está localizado o gene da distrofina; as meninas com uma variante causadora da doença têm uma cópia de segurança. Substituir até mesmo algumas proteínas de trabalho nas células musculares deve reverter a doença ou, pelo menos, interromper sua progressão.
Mas desenvolver essa substituição provou ser difícil. A distrofina é o gene mais longo do genoma humano e é muito grande para caber no vetor do vírus adeno-associado (AAV) comumente usado para fornecer terapias genéticas. A Sarepta e várias outras empresas contornaram isso projetando um gene que codifica apenas as partes mais importantes da proteína (consulte ‘Grandes genes em pacotes pequenos’). A “microdistrofina” resultante é apenas parcialmente eficaz.
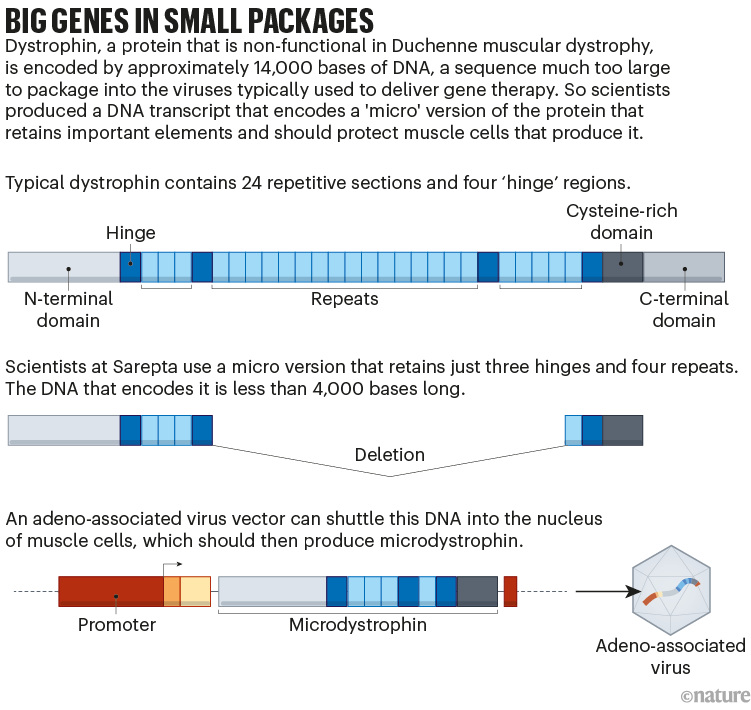
Relatórios da organização de notícias STAT e outros descobriram que a equipe da FDA planejava rejeitar o pedido de Sarepta. Mas Peter Marks, diretor do Centro de Avaliação e Pesquisa Biológica da FDA (CBER) interveio – agendando uma reunião pública com um comitê consultivo científico independente em 12 de maio, para avaliar se o medicamento deveria receber uma aprovação acelerada. Este comitê recomendou estreitamente a aprovação por oito votos a seis. Embora o FDA não precise seguir o conselho do comitê, ele normalmente o faz.
O comitê consultivo avaliou evidências de marcadores substitutos, bem como de benefícios clínicos. Alguns dos que votaram a favor da terapia dizem que, como a DMD progride lentamente, pode ser difícil determinar o efeito da droga depois de apenas um ano. Mas quando Sarepta olhou apenas para os 32 meninos com menos de seis anos de idade que receberam SRP-9001 ou um placebo, a função muscular dos meninos tratados melhorou significativamente.
A diretora científica da Sarepta, Louise Rodino-Klapac, não acha que a droga realmente funcione melhor nesses meninos mais jovens. Em vez disso, diz ela, os meninos mais velhos no grupo de controle do placebo eram mais saudáveis no início do estudo do que os mais jovens, fazendo com que os efeitos da droga parecessem mais fracos nesse grupo em comparação. Rodino-Klapac espera que o estudo de fase III em andamento, que melhor controlou a condição inicial de cada paciente, mostre que funciona igualmente bem nos músculos de meninos mais velhos.
A melhora estatisticamente significativa nos meninos mais jovens foi suficiente para convencer Donald Kohn, um membro do comitê consultivo da FDA que faz pesquisas com células-tronco na Universidade da Califórnia em Los Angeles, a votar a favor da droga. “Do ponto de vista estatístico, [Sarepta] não defendeu seu caso”, diz ele. Mas, acrescenta, a eficácia em meninos mais jovens sugere que a droga está tendo um efeito real. “É um voto de esperança”, diz Kohn.
Outros estão mais preocupados com o que parece ser uma falta geral de eficácia. “Entendo o apelo de ‘algo é melhor do que nada’, mas a questão é a que custo”, diz Caleb Alexander, epidemiologista da Universidade Johns Hopkins, em Baltimore, que também foi membro do comitê e votou contra o medicamento. Não apenas o SRP-9001 pode ter efeitos colaterais significativos, diz ele, mas os ensaios controlados por placebo concluídos até agora não sugerem fortemente que altos níveis de microdistrofina preveem resultados clínicos. “Nenhuma análise post-hoc de subgrupos de meninos muda esse fato”, diz ele.
A FDA e os membros do comitê consultivo também levantaram preocupações sobre se o Sarepta terminaria seu ensaio clínico, que ainda está avaliando mais de 60 meninos que inicialmente receberam o placebo e agora estão recebendo o medicamento. Eles apontaram que a empresa não completou os testes de três drogas DMD anteriores que o FDA havia aprovado de forma acelerada. Ainda estão no mercado. Um porta-voz de Sarepta diz que esses ensaios já inscreveram pacientes suficientes e estão em andamento.
“É uma decisão muito complexa. Não invejo o FDA”, diz Sharon Hesterlee, diretora de pesquisa da Muscular Dystrophy Association em Chicago, Illinois. Embora ela concorde que o efeito da terapia pode ser modesto e acarreta riscos significativos, “o risco de não fazer nada é 100% fatal”.
Quais são os riscos da terapia genética para DMD?
Além dos riscos das grandes quantidades de AAV necessárias para colocar o gene da microdistrofina nos músculos, o próprio gene parece causar efeitos colaterais graves em algumas crianças. A FDA suspendeu vários ensaios de terapia genética DMD devido a preocupações de contaminação e pacientes que ficaram gravemente doentes. E em 2021, a agência interrompeu temporariamente um estudo da Pfizer, com sede em Nova York, que está testando sua própria versão da microdistrofina, após a morte de um paciente.
Para investigar o problema, Pfizer, Sarepta e Solid Biosciences, com sede em Charlestown, Massachusetts, decidiram reunir seus dados de microdistrofina. Seu estudo, que será publicado em breve no New England Journal of Medicine, descobriu que certas mutações no gene da distrofina fazem com que o sistema imunológico reconheça a microdistrofina como um invasor estranho e a ataque, causando inflamação perigosa no músculo e no coração. As empresas começaram a rastrear geneticamente os participantes de ensaios clínicos e excluir aqueles com as mutações relevantes – menos de 5% no caso de Sarepta – o que elimina o problema por enquanto. Mas Carsten Bönnemann, neurologista do Instituto Nacional de Distúrbios Neurológicos e Derrame em Bethesda, Maryland, que co-liderou o estudo, diz que as terapias genéticas eventualmente terão que ser adaptadas para ajudar esses pacientes.
Ainda assim, Hesterlee observa que mesmo aqueles que são elegíveis para a terapia recém-aprovada de Sarepta têm uma escolha difícil pela frente. As terapias genéticas AAV podem ser administradas apenas uma vez na vida de uma pessoa: uma vez que o sistema imunológico encontra um vetor de vírus, é provável que o ataque no futuro. E como o AAV é o vetor de escolha para a maioria das terapias genéticas, os pais de uma criança com DMD podem ter que escolher entre tratar seu filho agora, com a única terapia aprovada, ou esperar na esperança de que a Sarepta ou outra empresa lance algo melhor. no futuro. Isso significa permitir que a doença progrida e, potencialmente, estabilizá-la somente depois que os meninos tiverem perdido mais função muscular. “Alguns meses podem ser muito para as famílias”, diz Hesterlee.
Que outras terapias genéticas para DMD estão no horizonte?
A Pfizer está realizando um estudo de fase III em 99 meninos com sua versão da microdistrofina em um vetor AAV semelhante e espera divulgar seus resultados iniciais no próximo ano. Várias outras empresas estão desenvolvendo suas próprias microdistrofinas, incluindo a Regenxbio em Rockville, Maryland, que lançou um pequeno teste no início deste ano.
Outros estão procurando maneiras de melhorar o vetor viral que carrega o gene. Em 2022, a Solid Biosciences interrompeu seu teste de fase II de sua própria formulação de microdistrofina para mudar para um vetor diferente que visa especificamente as células musculares e provavelmente pode ser administrado em doses mais baixas. A empresa planeja começar a dosar pacientes ainda este ano. E na reunião anual da American Society for Gene & Cell Therapy em Los Angeles em maio, Chamberlain apresentou um novo sistema envolvendo vários vetores AAV, cada um carregando uma porção do gene da distrofina. Os produtos dos genes podem então se unir para criar uma versão da distrofina que é muito maior do que um único vetor poderia carregar. Mais adiante no horizonte estão abordagens que irão infundir pacientes com genes que codificam o sistema de edição do genoma CRISPR-Cas9 com a intenção de alterar permanentemente o próprio gene da distrofina nas células musculares.
E outros pesquisadores estão procurando vetores alternativos, incluindo cápsulas lipídicas, nanopartículas, anticorpos que carregam genes de distrofina para células musculares e vírus que se integram ao genoma de um paciente. Esses tratamentos, pelo menos em teoria, poderiam ser administrados a pessoas que já receberam terapia genética AAV.
Mas com uma terapia aprovada agora disponível, as empresas que tentam testar suas próprias terapias podem achar mais difícil recrutar participantes do estudo, que podem não querer correr o risco de receber um placebo. Além disso, a pesquisadora de serviços de saúde Reshma Ramachandran, da Yale University em New Haven, Connecticut, teme que, se as empresas souberem que podem obter a aprovação de uma terapia com microdistrofina, será mais provável que criem suas próprias versões ligeiramente aprimoradas, em vez de gastar mais tempo desenvolvendo suas próprias abordagens inovadoras.
Como essa aprovação afetará outras terapias genéticas?
O desenvolvimento da terapia genética sempre foi um negócio arriscado e caro, e a recente crise econômica fez com que muitas empresas de biotecnologia fechassem. No entanto, o número de aprovações de terapia genética pelo FDA aumentou a cada ano desde o primeiro – Kymriah para leucemia – em 2017.
A aprovação para uma terapia genética DMD ainda representaria um marco, no entanto. Até agora, a maioria das terapias genéticas aprovadas tratavam de cânceres, doenças e condições extremamente raras, como distúrbios da retina, que são fáceis de atingir com um vírus. A DMD é diferente, diz Chamberlain, tanto no número de desafios técnicos que os pesquisadores tiveram que enfrentar quanto no grande número de pacientes que a terapia poderia atender. A agência agora está pronta para considerar mais de uma dúzia de terapias genéticas e celulares este ano, incluindo duas para a doença falciforme – uma condição genética que é muito mais prevalente do que a DMD.
A FDA parece estar priorizando essas abordagens. Em conversas recentes, o diretor do CBER, Marks, disse que a agência planeja se apoiar mais fortemente nos caminhos de aprovação acelerada para terapias genéticas, incluindo o uso mais amplo de endpoints substitutos. “Não podemos ser tão cuidadosos com nossas aprovações sob aprovação acelerada a ponto de impedir que terapias potencialmente salvadoras cheguem ao mercado em tempo hábil”, disse ele em um discurso em janeiro na reunião anual da Associação de Distrofia Muscular.
Isso preocupa alguns cientistas, que temem que os pacientes acabem pagando preços altos por medicamentos ineficazes. “Acho que a noção de flexibilidade regulatória pode ficar fora de forma”, diz Alexander. Ramachandran aponta para a aprovação acelerada do FDA em 2021 de um medicamento para Alzheimer, o aducanumab, que eliminou com sucesso as placas amiloides no cérebro dos pacientes, mas não melhorou de forma mensurável sua saúde. Posteriormente, o FDA aprovou outro medicamento semelhante que mostrou apenas um benefício clínico menor. “Pacientes com a doença de Duchenne não merecem apenas mais opções, eles precisam de melhores opções”, diz Ramachandran.
Mas Chamberlain afirma que mesmo um pequeno sucesso ajudará todo o campo. “Há um enorme espaço para melhorias”, diz ele. “Acho que isso vai nos dar algum tempo para fazer essas melhorias.”
Publicado em 07/06/2023 20h36
Artigo original: