


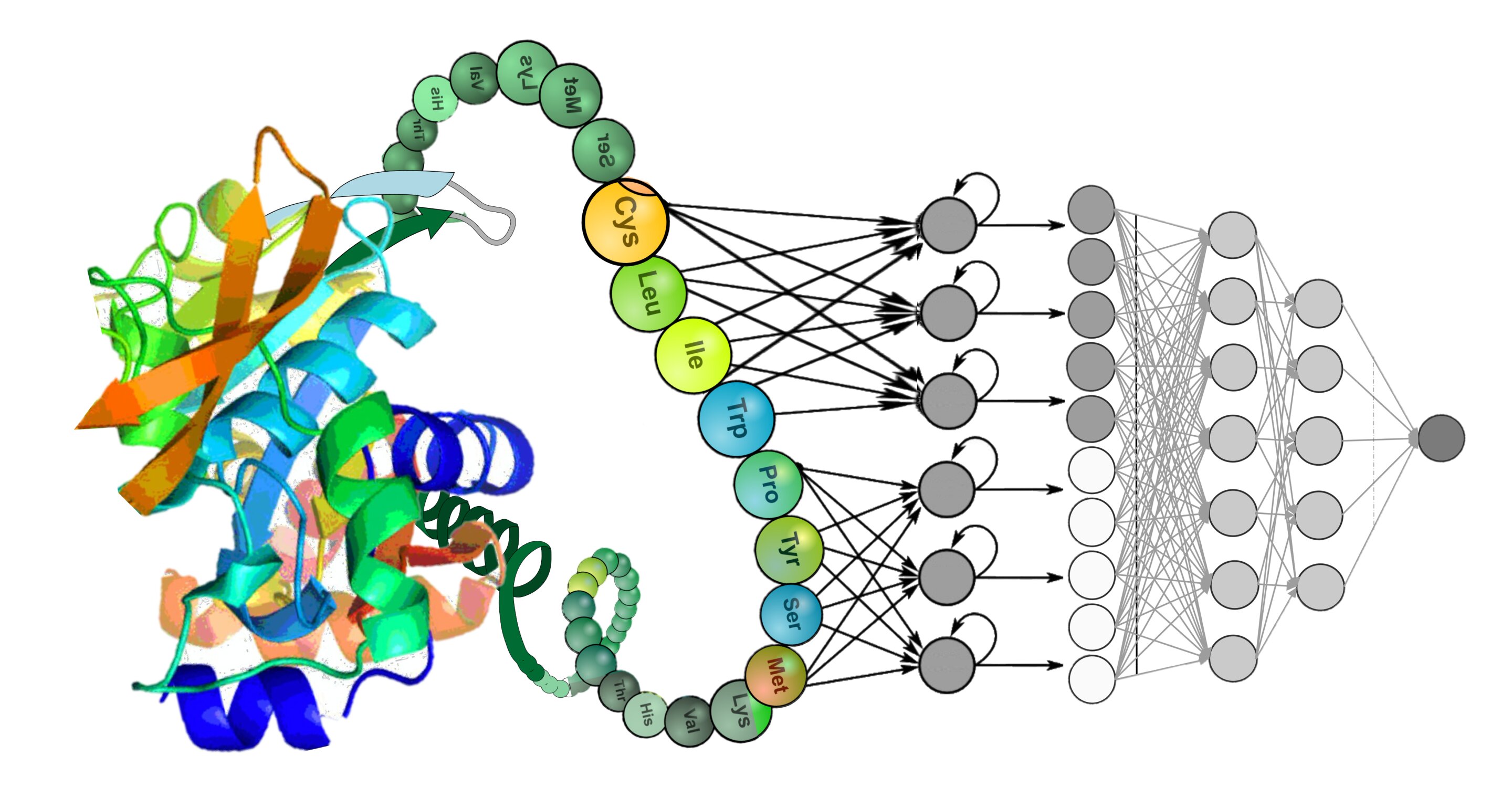
Modelos de linguagem baseados em aprendizado profundo, como BERT, T5, XLNet e GPT, são promissores para analisar fala e textos. Nos últimos anos, no entanto, eles também têm sido aplicados nas áreas de biomedicina e biotecnologia para estudar códigos genéticos e proteínas.
Bioinformáticos, pesquisadores de genética e neurocientistas vêm tentando inferir os papéis biológicos de genes e proteínas há décadas. Para fazer isso, no entanto, eles precisam analisar dados biológicos extremamente grandes e altamente complexos.
Pesquisadores da Hacettepe University, Middle East Technical University e Karadeniz Technical University, Turquia, realizaram recentemente um estudo avaliando o potencial de modelos de linguagem baseados em aprendizado profundo para estudar proteínas e prever suas propriedades funcionais. Seu artigo, publicado na Nature Machine Intelligence, fornece um resumo valioso das vantagens e desvantagens de diferentes abordagens de última geração.
“Dados de biologia molecular podem ser modelados como uma linguagem (ou seja, a linguagem de genes/proteínas), de modo que a sequência de um gene ou proteína possa ser pensada como uma frase com um significado específico em linguagem natural, e a semântica dessa linguagem protéica são as propriedades biológicas, físicas e químicas específicas dessas biomoléculas”, disse Tunca Dogan, um dos pesquisadores que realizaram o estudo, ao Phys.org. “Com base nessa ideia, nosso trabalho tenta construir modelos de aprendizado de máquina que usam incorporações numéricas de proteínas de alta dimensão derivadas de modelos de linguagem como entrada e predizem suas propriedades funcionais com alta precisão”.
Em seu artigo, Dogan e seus colegas avaliaram a capacidade de diferentes abordagens de modelagem de linguagem de proteínas para extrair padrões ocultos contendo pistas importantes sobre as propriedades funcionais das proteínas. Suas avaliações incluíram todas as arquiteturas de modelagem de linguagem natural mais conhecidas (ou seja, BERT, T5, XLNet, ELMO, etc.), cada uma das quais pode conter centenas de milhões ou, em alguns casos, bilhões de parâmetros.
“O pré-treinamento autossupervisionado desses modelos requer enormes recursos”, explicou Dogan. “Graças ao valioso trabalho anterior sobre este tópico, que visava pré-treinar modelos de linguagem de proteínas usando essas arquiteturas, focamos principalmente em nosso treinamento secundário supervisionado para prever propriedades funcionais”.
Para avaliar efetivamente os modelos de linguagem de proteínas e comparar seus desempenhos, a equipe primeiro teve que compilar conjuntos de dados de teste grandes e confiáveis, cada um com um nível de dificuldade diferente. Por fim, eles criaram quatro conjuntos de dados de referência que permitiram investigar semelhanças semânticas, definições funcionais baseadas em ontologia, famílias de proteínas alvo de drogas e interações físicas entre proteínas. Todos esses são mecanismos biológicos cruciais que são conhecidos por estarem intimamente ligados à ocorrência e progressão de doenças geneticamente herdadas, como diferentes tipos de câncer.
“Talvez nossa descoberta mais notável tenha sido que esses modelos de linguagem profunda são capazes de aprender com sucesso as propriedades funcionais das proteínas usando as sequências de aminoácidos como única entrada, o que é um problema bastante difícil”, disse Dogan. “Esses resultados também são consistentes com as descobertas de estudos recentes de previsão da estrutura da proteína (por exemplo, AlphaFold2 da Deepmind e RoseTTAFold da Baker Lab), que usa a sequência como entrada e prevê a estrutura do monômero 3D com desempenho extremamente alto”.
No futuro, os modelos avaliados por essa equipe de pesquisadores poderão ajudar a aprimorar as intervenções de medicina de precisão, por exemplo, analisando a composição molecular de pacientes resultantes de variações genômicas para elaborar tratamentos personalizados. Embora os resultados reunidos por Dogan e seus colegas destaquem o enorme potencial das ferramentas de modelagem de proteínas baseadas em aprendizado profundo, os métodos existentes ainda precisarão ser significativamente aprimorados antes que possam ser integrados aos sistemas de tomada de decisão clínica da vida real.
“Agora estamos trabalhando em um novo sistema para representar melhor as proteínas”, acrescentou Dogan. “Além das sequências de aminoácidos, este sistema utiliza dados baseados em rede (ou seja, interações proteína-proteína conhecidas) e conhecimento oculto nos textos biomédicos não estruturados (por exemplo, artigos científicos) no nível de entrada, juntamente com abordagens integrativas de aprendizado profundo. Nosso objetivo final é obter uma representação universal de proteínas que possa ser usada com sucesso em qualquer tarefa de modelagem biomédica ou biotecnológica.”
Publicado em 21/04/2022 09h23
Artigo original:
Estudo original: