



A infecção dos vírus da imunologia humana (HIV) ocorre pela integração de seu genoma em células infectadas para entrar em um estado inativo de latência reversível que escapa à terapia antirretroviral. A capacidade de sequenciar tal provírus e junções hospedeiras adjacentes em células individuais pode destacar seu mecanismo de ação e persistência em células infectadas.
No entanto, este experimento é difícil de realizar devido à quantidade 150 milhões de vezes maior de DNA humano de fundo. Em um novo relatório agora publicado na Nature Biomedical Engineering, Chen Sun e uma equipe de pesquisa em bioengenharia e persistência viral e estudos de dinâmica na Universidade da Califórnia, San Francisco, NIH e no Chan Zuckerberg Biohub, nos EUA, mostraram como os provírus são conectados às junções de DNA do hospedeiro HIV por meio de ensaios microfluídicos de alto rendimento, para amplificação do genoma inteiro baseado em gotículas do DNA do HIV em seu contexto nativo. A equipe conduziu uma reação em cadeia da polimerase (PCR) para marcar gotículas contendo provírus para sequenciar e testar células infectadas de pessoas com HIV, recebendo terapia antirretroviral supressora no momento para detectar e sequenciar genomas provirais pareados e locais de integração. O trabalho procurou melhorar a análise genética de reservatórios persistentes de células infectadas pelo HIV.
O estado atual do tratamento do HIV e estratégias terapêuticas em evolução
Enquanto aproximadamente 37 milhões de pessoas em todo o mundo estão infectadas com o vírus da imunodeficiência humana (HIV), a cura permanece desconhecida. O mecanismo de infecção do vírus impõe um obstáculo central e também detém a chave para a cura da doença, pois o HIV integra seu genoma ao genoma das células infectadas. Embora a terapia antirretroviral (ART) possa suprimir a replicação do vírus a níveis indetectáveis e prevenir a progressão da síndrome da imunodeficiência adquirida (AIDS), ela não possui atividade contra reservatórios celulares. Como resultado, a terapia deve ser continuada indefinidamente para evitar o rebote do vírus e a progressão da doença. É difícil lidar com o estigma associado ao HIV, devido ao custo da administração de TARV ao longo da vida, com potencial tóxico. Portanto, é importante erradicar ou suprimir os reservatórios celulares infectados pelo HIV para obter uma cura funcional. Para examinar a persistência de reservatórios celulares, os cientistas devem realizar análises pareadas de locais de integração e sequências de provírus de comprimento total. Ao estabelecer um método que isola e sequencia de forma confiável e econômica os pró-vírus raros, os pesquisadores podem caracterizar de forma abrangente os reservatórios de HIV. Sun et al desenvolveram tal abordagem para caracterizar provírus e seu contexto genômico celular dentro de reservatórios de HIV usando local de integração simultânea e sequenciamento de provírus (SIP-Seq).
Superando as probabilidades: um método para recuperar genomas de provírus únicos
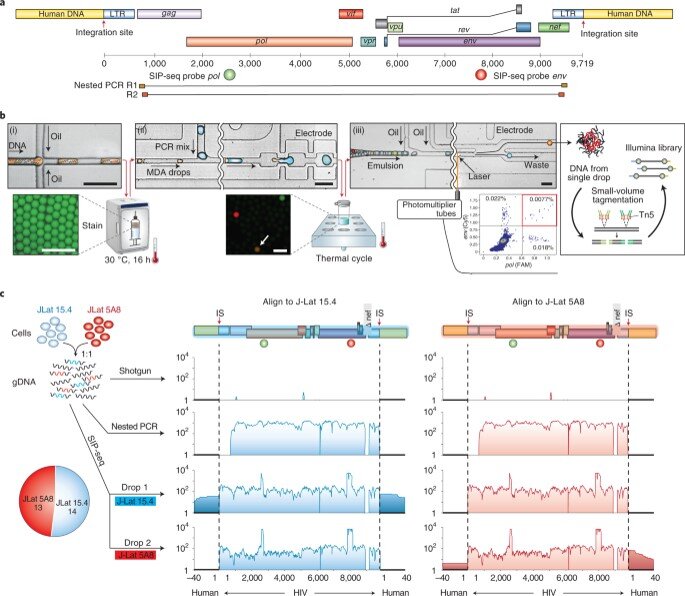
Usando amplificação de sequenciamento de genoma completo, Sun et al amplificaram o genoma do HIV em seu contexto nativo dentro de gotículas microfluídicas, então marcaram as gotículas contendo provírus para sequenciamento. O método resultante forneceu um genoma de vírus completo conectado às suas junções de célula hospedeira por meio de um único conjunto contínuo. A velocidade e a eficiência do método permitiram a recuperação de genomas de provírus únicos em um fundo de DNA 150 milhões de vezes maior. A equipe usou SIP-Seq (local de integração simultânea e sequenciamento de provírus) para traçar o perfil da população de provírus em uma variedade de indivíduos relacionados ao ART para entender o reservatório latente do HIV. Enquanto a equipe se concentrou no HIV, essa abordagem é aplicável a diversos vírus que invadem o genoma do hospedeiro, para fornecer uma plataforma geral para caracterizar a genética de uma variedade de infecções.

Os experimentos
Sun et al buscaram recuperar todos os fragmentos de DNA contendo genomas de HIV de uma população de milhões de células T CD4+, para sequenciar as moléculas individualmente e suas junções imediatamente adjacentes. A equipe extraiu grandes fragmentos de DNA de células encapsuladas em microgotas para isolar os genomas integrados do HIV. Usando amplificação de deslocamento multiplexado (MDA), eles amplificaram não especificamente cada fragmento genômico para obter provírus únicos suficientes para sequenciamento. Eles então seguiram isso isolando gotículas individuais com provírus de HIV, onde cada gotícula foi codificada em barras e sequenciada. Os virologistas então mapearam as leituras de cada gotícula para um genoma de referência do HIV para identificar os locais de integração para facilitar informações claras sobre a complexidade do genoma do vírus e seus locais de integração – cruciais para avaliar o reservatório celular.

Detectando provírus com SIP-Seq e regulando microfluídica
Para aumentar a especificidade de provírus de comprimento total e prevenir a clivagem aleatória do DNA, Sun et al empregaram uma PCR de Taqman multiplexada específica dupla, para atingir regiões no genoma do HIV. Durante os experimentos, eles produziram um volume específico de DNA para sequenciar e confirmar a recuperação do provírus HIV. A microfluídica da configuração SIP-Seq continha três dispositivos, incluindo um encapsulador de gotículas, uma fusão e um classificador. Os cientistas carregaram os fragmentos de DNA genômico humano com reagentes de amplificação de deslocamento múltiplo (MDA) para encapsular até 10 bilhões de fragmentos de DNA, com aproximadamente 75 kbp de comprimento, em 20 milhões de gotículas separadas, em 15 minutos. Cada gota continha 500 fragmentos distintos e Sun et al os incubaram a 30 graus Celsius, para amplificação não específica, antes da fusão com reagentes Taqman PCR. Depois de repetir as etapas para todos os genomas de HIV detectados, a equipe preparou os tubos para sequenciamento.

Analisando os provírus do HIV e seu cenário genômico em pessoas tratadas com TARV
Sun et al validaram o SIP-Seq em linhas de células de HIV e analisaram o HIV em pessoas tratadas com ART. Durante os experimentos, eles prepararam células por plaqueamento, células T CD4+ em repouso de uma pessoa tratada com ART, seguido de estimulação e cultura in vitro para proliferação por meio de três métodos; shotgun, PCR aninhado e SIP-Seq com sondas Taqman. Tanto o SIP-Seq quanto o nested PCR produziram excelentes resultados, a equipe optou especificamente pelo SIP-Seq para caracterizar toda a diversidade genética das células infectadas in vivo. Eles seguiram esse trabalho caracterizando a paisagem genômica dos provírus do HIV, que isolaram diretamente das células T CD4+ de pessoas infectadas. Os resultados revelaram três linhagens clonais para apoiar a origem clonal em relação a estudos anteriores dos mesmos indivíduos.
Panorama
A pesquisa indicou como os métodos permitiram que Chen Sun e seus colegas estudassem os provírus do HIV de forma mais eficiente do que com os métodos de PCR existentes. Os resultados forneceram uma análise abrangente do cenário genético do HIV in vivo, caracterizando o reservatório latente para destacar seu papel na persistência do HIV. Os métodos existentes eram desafiadores devido à raridade e falta de marcadores de superfície distintos para identificar células infectadas de forma latente. Usando o método descrito SIP-Seq (simultaneous integration site and provirus sequencing), Sun et al forneceram um processo rápido, econômico e escalável, que aplicaram a pessoas recebendo terapia antirretroviral (ART), para destacar a presença em reservatórios latentes de células expandidas clonalmente. A estratégia é aplicável em infecções por HIV, a outras doenças virais com estados integrados em seu ciclo de vida.
Publicado em 14/04/2022 06h49
Artigo original:
Estudo original: