


Os cientistas esperaram meses pelo acesso a uma previsão de estrutura de proteína altamente precisa desde que a DeepMind apresentou um progresso notável nesta área na Conferência de Avaliação Crítica de Predição de Estrutura de 2020, ou CASP14. A espera agora acabou.
Pesquisadores do Institute for Protein Design da University of Washington School of Medicine em Seattle, em grande parte, recriaram o desempenho alcançado pela DeepMind nesta importante tarefa. Esses resultados serão publicados online pela revista Science na quinta-feira, 15 de julho.
Ao contrário do DeepMind, o método da equipe UW Medicine, que eles apelidaram de RoseTTAFold, está disponível gratuitamente. Cientistas de todo o mundo agora estão usando-o para construir modelos de proteínas para acelerar suas próprias pesquisas. Desde julho, o programa foi baixado do GitHub por mais de 140 equipes de pesquisa independentes.
As proteínas consistem em cadeias de aminoácidos que se dobram em intrincadas formas microscópicas. Essas formas únicas, por sua vez, dão origem a quase todos os processos químicos dentro dos organismos vivos. Ao compreender melhor as formas das proteínas, os cientistas podem acelerar o desenvolvimento de novos tratamentos para o câncer, COVID-19 e milhares de outros distúrbios de saúde.
“Tem sido um ano agitado no Institute for Protein Design, projetando terapêuticas e vacinas COVID-19 e lançando-as em ensaios clínicos, juntamente com o desenvolvimento de RoseTTAFold para previsão de estrutura de proteína de alta precisão. Estou muito satisfeito que a comunidade científica já esteja usando o RoseTTAFold servidor para resolver problemas biológicos pendentes”, disse o autor sênior David Baker, professor de bioquímica da Escola de Medicina da Universidade de Washington, investigador do Howard Hughes Medical Institute e diretor do Institute for Protein Design.
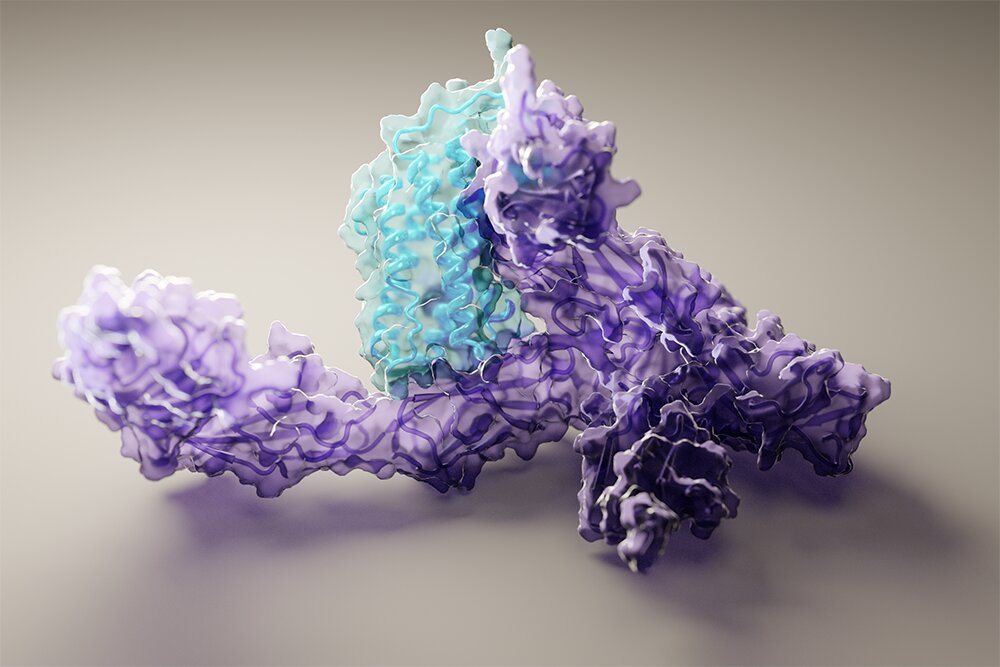
No novo estudo, uma equipe de biólogos computacionais liderada por Baker desenvolveu a ferramenta de software RoseTTAFold. Ele usa o aprendizado profundo para prever com rapidez e precisão as estruturas das proteínas com base em informações limitadas. Sem a ajuda desse software, pode levar anos de trabalho de laboratório para determinar a estrutura de apenas uma proteína.
RoseTTAFold, por outro lado, pode computar de forma confiável uma estrutura de proteína em apenas dez minutos em um único computador de jogo.
A equipe usou RoseTTAFold para calcular centenas de novas estruturas de proteínas, incluindo muitas proteínas mal compreendidas do genoma humano. Eles também geraram estruturas diretamente relevantes para a saúde humana, incluindo aquelas para proteínas associadas ao metabolismo lipídico problemático, distúrbios de inflamação e crescimento de células cancerosas. E eles mostram que RoseTTAFold pode ser usado para construir modelos de conjuntos biológicos complexos em uma fração do tempo anteriormente necessário.
RoseTTAFold é uma rede neural de “três vias”, o que significa que considera simultaneamente padrões nas sequências de proteínas, como os aminoácidos de uma proteína interagem entre si e a possível estrutura tridimensional de uma proteína. Nessa arquitetura, as informações unidimensionais, bidimensionais e tridimensionais fluem para frente e para trás, permitindo assim que a rede raciocine coletivamente sobre a relação entre as partes químicas de uma proteína e sua estrutura dobrada.
“Esperamos que esta nova ferramenta continue a beneficiar toda a comunidade de pesquisa”, disse Minkyung Baek, um pós-doutorado que liderou o projeto no laboratório Baker na UW Medicine.
Publicado em 18/07/2021 16h15
Artigo original:
Estudo original: